This article is now archived.
WDL articles now live in the openwdl/wdl-docs GitHub repository. Find the updated documentation on the wdl-docs website.
The ability to connect outputs to inputs described in Linear Chaining, which relies on hierarchical naming, allows you to chain together tools that produce multiple outputs and accept multiple inputs, and specify exactly which output feeds into which input.
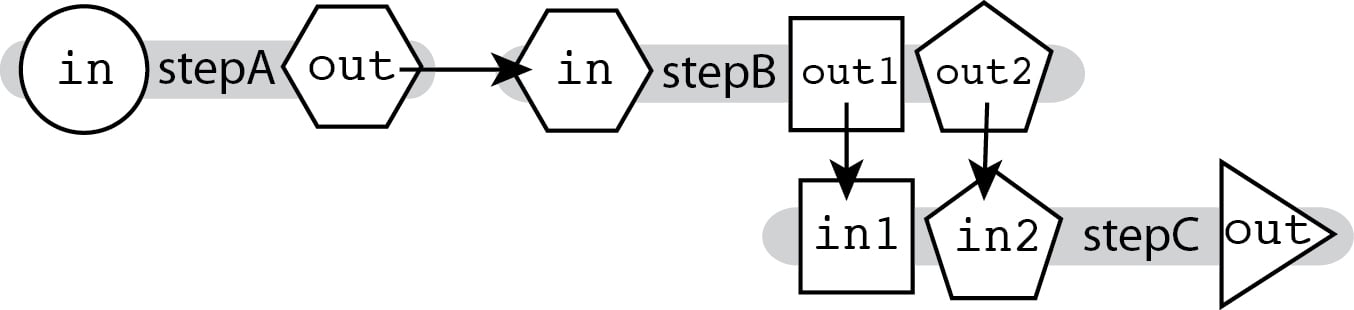
As the outputs for stepB are named differently, we can specify where exactly each output goes in the next step's input fields.
call stepC { input: in1=stepB.out1, in2=stepB.out2 }
Generic example script
In context, this sort of plumbing would look as follows:
workflow MultiOutMultiIn {
File firstInput
call stepA { input: in=firstInput }
call stepB { input: in=stepA.out }
call stepC { input: in1=stepB.out1, in2=stepB.out2 }
}
task stepA {
File in
command { programA I=${in} O=outputA.ext }
output { File out = "outputA.ext" }
}
task stepB {
File in
command { programB I=${in} O1=outputB1.ext O2=outputB2.ext }
output {
File out1 = "outputB1.ext"
File out2 = "outputB2.ext" }
}
task stepC {
File in1
File in2
command { programB I1=${in1} I2=${in2} O=outputC.ext }
output { File out = "outputC.ext" }
}
Concrete example
This workflow uses Picard’s splitVcfs tool to separate the original VCF into two VCF files, one containing only SNPs and the other containing only Indels. The next step takes those two separated VCFs and recombines them into one.
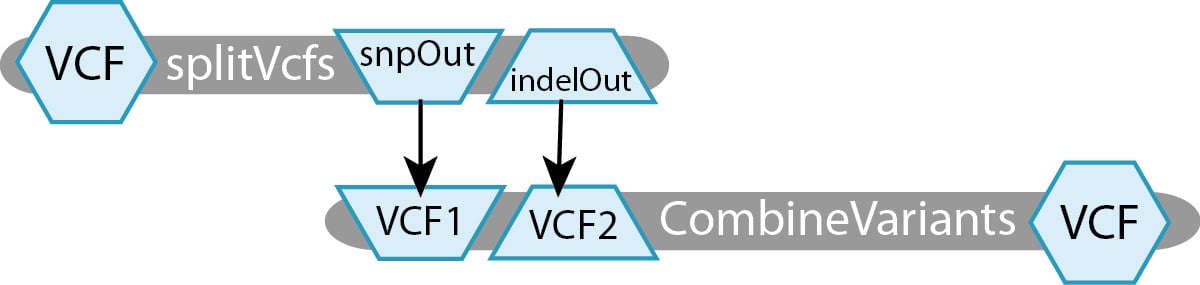
For this toy example, we have defined two tasks:
- splitVcfs takes in a
File VCFand outputs aFile snpOutand aFile indelOut. - CombineVariants takes in a
File VCF1and aFile VCF2, producing aFile VCF.
Concrete example script
The workflow described above, in its entirety, would look like this:
workflow MultiOutMultiInExample {
File inputVCF
call splitVcfs { input: VCF=inputVCF }
call CombineVariants { input: VCF1=splitVcfs.indelOut, VCF2=splitVcfs.snpOut }
}
task splitVcfs {
File VCF
command {
java -jar picard.jar SplitVcfs \
I=${VCF} \
SNP_OUTPUT=snp.vcf \
INDEL_OUTPUT=indel.vcf
}
output {
File snpOut = "snp.vcf"
File indelOut = "indel.vcf"
}
}
task CombineVariants {
File VCF1
File VCF2
command {
java -jar GenomeAnalysisTK.jar \
-T CombineVariants
-R reference.fasta \
-V ${VCF1} \
-V ${VCF2} \
--genotypemergeoption UNSORTED \
-o combined.vcf
}
output {
File VCF = "combined.vcf"
}
}
Note that here for simplicity we omitted the handling of index files, which has to be done explicitly in WDL. For examples of how to do that, see the Tutorials.