This article explains what entities are, and some of the more technical details of Terra's five default root entity types - part of the standard genomic data model.
Entity overview: What's an entity?
If you're new to Terra, you may first see reference to "entity types" in a workflow configuration form.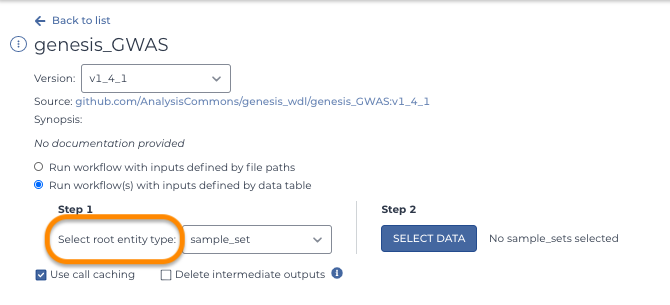
The "root entity" is the smallest piece of data a workflow can use as input and the "root entity type" is the table containing that root entity as it's primary data (such as a sample or specimen, or a sample_set or specimen_set). You select the root entity type of input when setting up a workflow. (see Workflow setup part 1: Runtime options).
How does an entity relate to your data? According to the dictionary, an "entity" is "a thing with distinct and independent existence." In Terra, an "entity" is the primary type of data stored in a data table: also the table in the workspace Data page.
You can have tables of sample data (a "sample" entity table) or tissues (a "tissue" table), or unicorns (a "unicorn" table), for example. The combined entities in your workspace - and their relationships to each other - make up your data model.
Example entity: sample
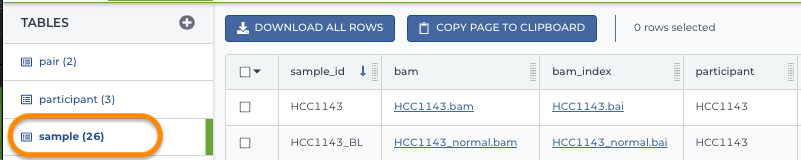
This sample table includes genomic data (BAM and BAM index files) of various samples. Note that the first column is each sample's unique ID and the fourth column is the participant ID, also found in the participant table.
Example entity: fruit
Terra has a flexible data model that allows you to define any entity you want. For example, you can have a "fruit" table such as this one (say you're doing research on the genomes of different varieties of apples and oranges).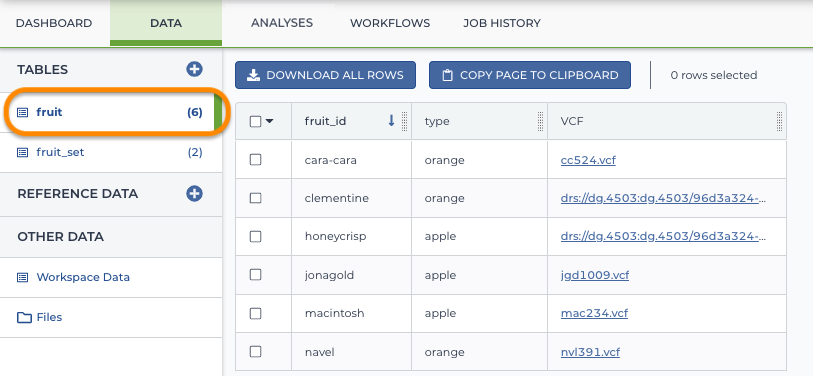
Example entity: fruit_set
Your workflow might take an array of samples from a particular fruit, or you might want to want to run separate analyses on apples and oranges.In these cases, you would create an "apple" set and an "orange" set using an "entity_set" membership table (in this case a "fruit_set" table). The fuit_set table includes the IDs for apples and oranges in each set. The individual fruit IDs are from the "fruit" table (above).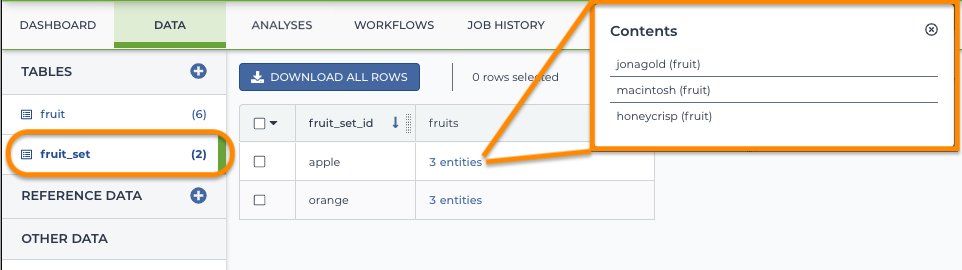
Default entities: Terra's standard genomic data model
In Terra, five default root entity types are predefined with specific relationships. These make up the "standard genomic data model" - originally developed based on the types of data data typically used in cancer research. Using default entity names helps standardize workflows where the end result depends on grouping and associating the data properly.
The standard genomic data model - default entity types
Terra's standard genomic data model is made up of a number of nested tables of particular entities.
Participant | Participant set | Sample | Sample set | Pair | Pair set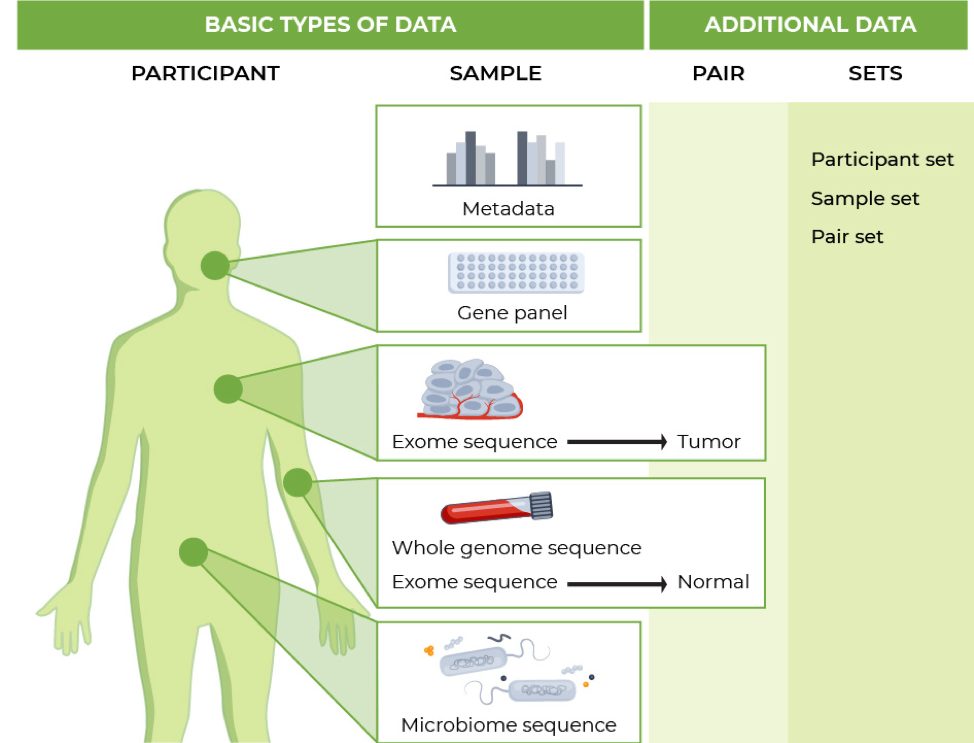
Participant: An individual, such as someone in a study. Participant IDs help keep track of samples that belong to the same individual. They're useful, for instance, if survey subjects provide multiple types of biological samples (spit, blood, etc).
Participant set: A grouping of particular participants. This entity type can be multiply nested, as it can group multiple participants, each with multiple samples.
Sample: The most basic genomic data entity is the nucleotide sequence from a single lane.
Sample set: A grouping of particular samples. You can use this entity type for running a single-input workflow on the same samples over and over.You can also use this nested entity type in workflows that require arrays (multiple samples) as input, such as joint genotyping or panel-of-normals (PoN) generation.
Pair: A set of paired tumor and normal samples taken from the same patient. This entity type is tailored for somatic workflows.
Pair set: Sets of tumor-normal pairs. For example, a study may identify several tumor-normal pairs taken from the same or different participants.
Origins of the default entity types (standard genomic data model) in Terra Terra's five default entity types are not native to the Workflow Description Language (WDL). The concept of entity types in Terra/GATK is inherited from TCGA's metadata style, which was designed to categorize different types of data files typically used in cancer research - such as files that track treatment regimens, files that track the progress of samples through pipelines, etc. Using the same entity names and default relationships made it easier to standardize workflows for similar sorts of analyses.
Terra now uses flexible entity types
The analyses you can do on the Terra platform has expanded beyond cancer research and you can use any entity description you want. However, you can take advantage of these predefined relationships by using these default root entity types.
Default entity types streamline linking data in different tables
In a table, each distinct entity (sample or specimen or participant) has its own row - specified by its own unique ID. However, data in different tables are often related, such as a single participant's phenotypic data and genomic data (often found in the participant and sample tables, respectively).
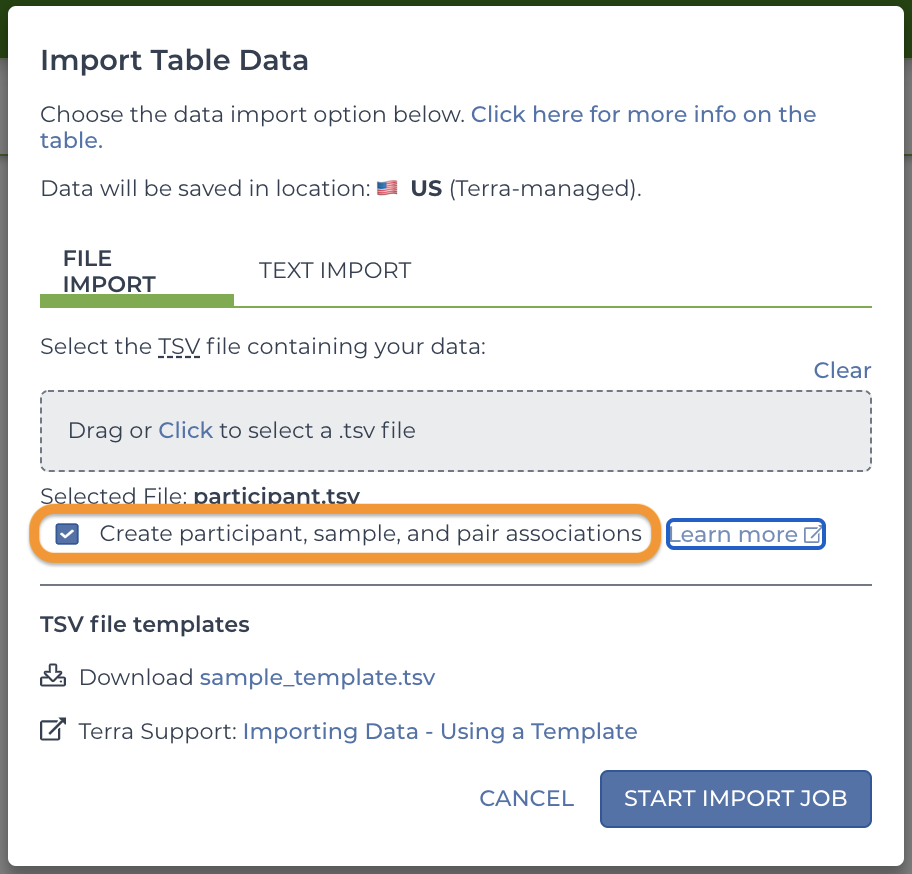
How to connect some types of data automaticallyTerra can associate some types of data automatically (which helps streamline workflows that expect associated data). To do so, you must make sure to do all of the following.
1. Use the default entity types (the "standard genomic data model").
2. Upload tables in a specific order (participants > sample > pair).
3. Check "Create participant, sample, and pair associations" in the Import Table Data form.
General upload order rule
Upload tables that reference other tables after the tables they reference. For example, you must upload the sample table before uploading a sample_set table (that references the sample_id from the sample table). See how to associate participants, samples, and pairs (and sets of these) in the examples below.
Example: How to link study participants to their genomic data
1. Upload the participant table
Imagine a study with thousands of participants. You define each participants' unique ID in a participant table. The first column is the participant ID. Additional columns include other information associated with the participant. Note that traditionally a participant table only included participant IDs, but today a participant table might also include other data related to the participant, such as phenotypic data (as in the screenshot below):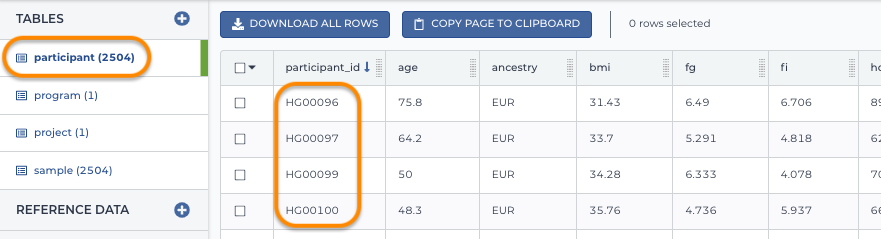
2. Upload the sample table
Traditionally a sample table referenced genomic data files in cloud storage plus additional data. The sample entity table shown below includes 1) the unique ID for each sample, 2) links to the sample data files in the cloud, and 3) the participant_ID (from the participant table). The participant ID links the sample to the participant table.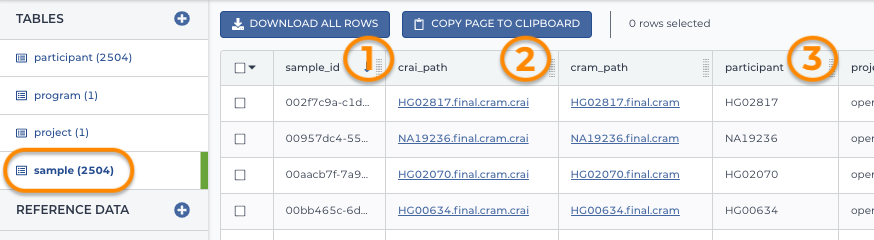
Example: How to group individual entities into sets
In this example, there are both mouse and human specimens in the specimen table. It can be useful to make a set of just the mouse samples for analysis.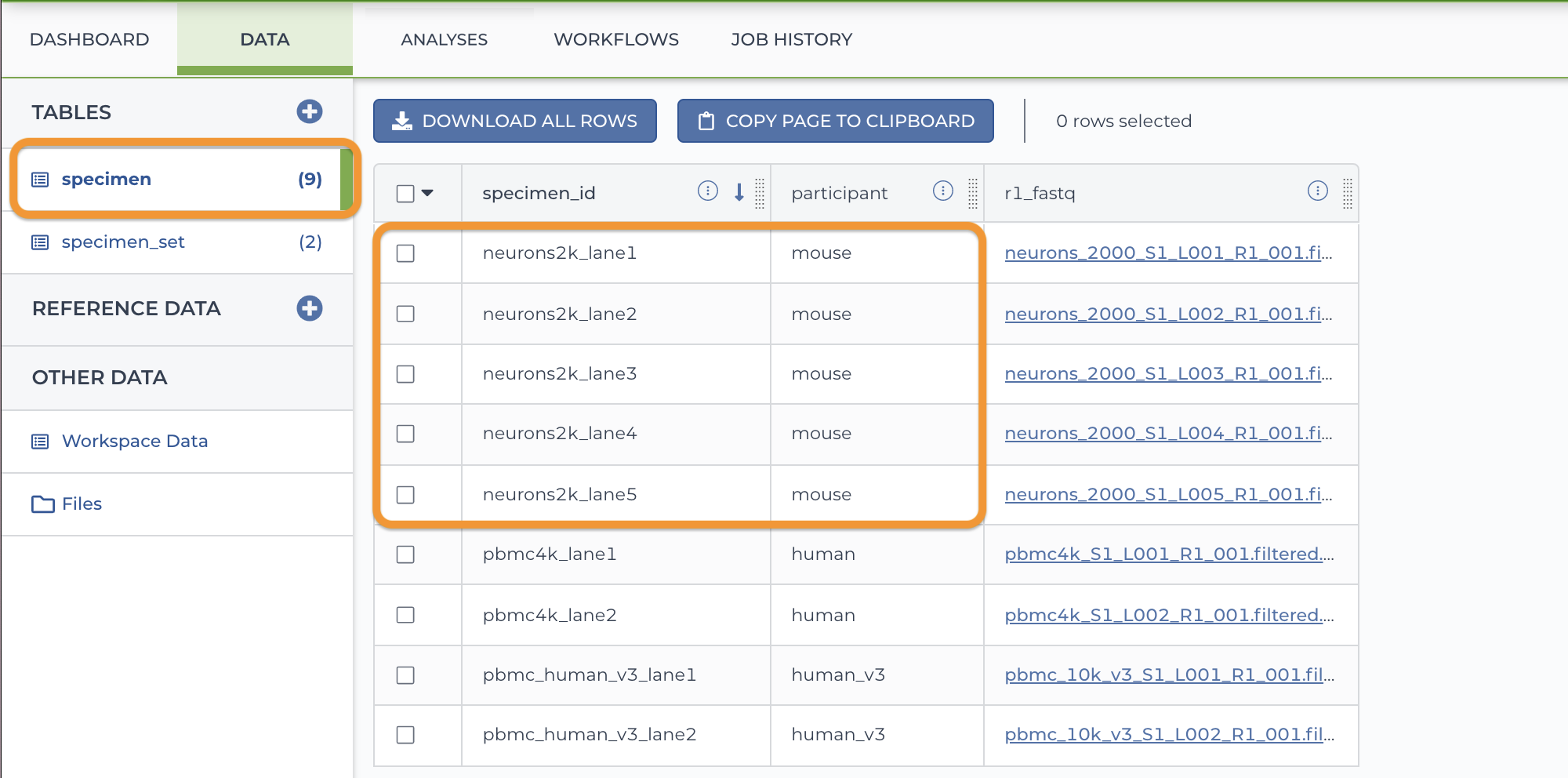
1. Load the specimen table.
2. Load the specimen_set entity (table). In the screenshot below, the set "mouse" includes the five mouse specimens. 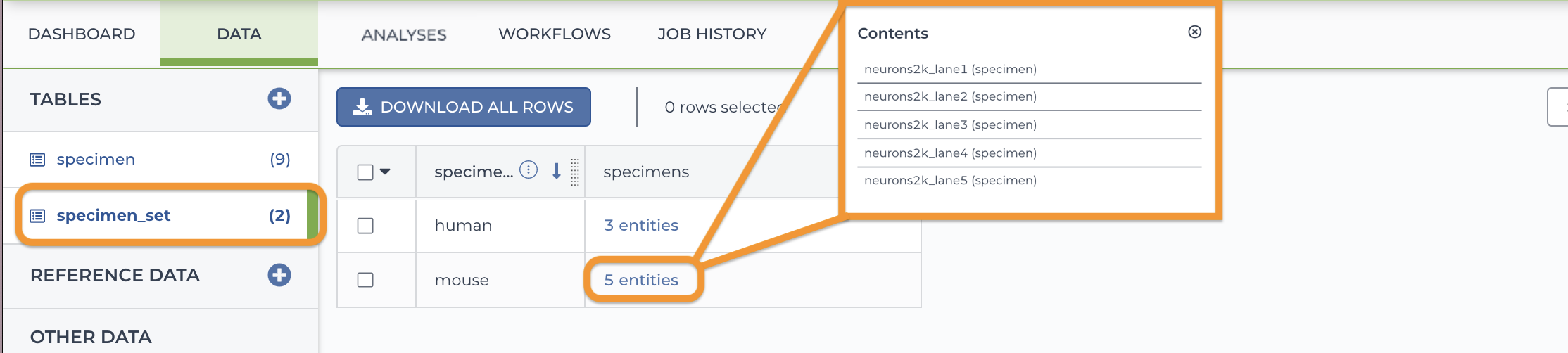
Linking data in participant, sample, and pair tables
Our Somatic Variant Discovery Best Practice guidelines stress the importance of comparing a matched normal sample to the tumor sample. Workflows that compare tumor tissue with normal tissue typically expect a pair set as the input entity. If you attempt to launch a Mutect2 workflow by selecting the tumor and the normal files as individual samples, the workflow will fail.
The tables you would include in your workspace are (in order of how they must be loaded): participant, sample, pair.
1. Load participant table
Notice this table doesn't reference any other table or entity.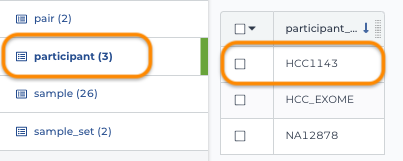
2. Load the sample table
Notice this references the participant_id from the first table.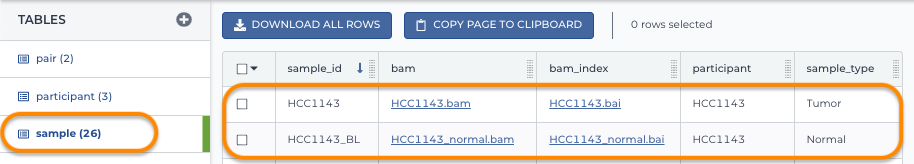
3. Load the pair table
Notice this references both the participant and sample IDs.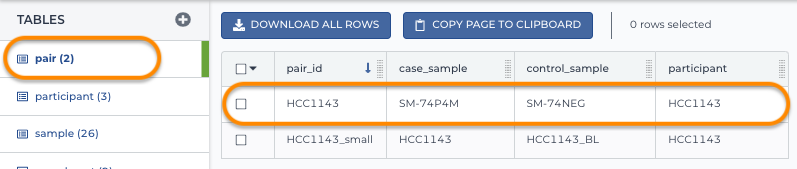
See the short video clip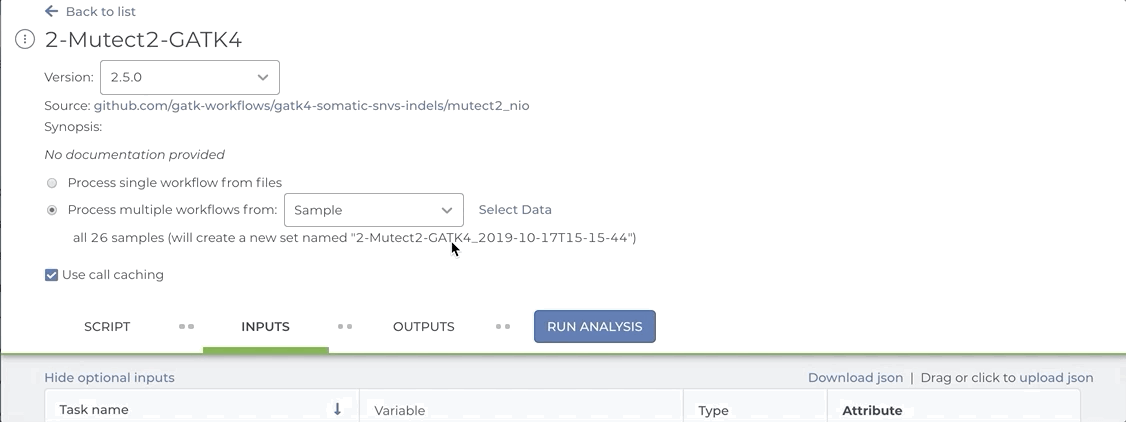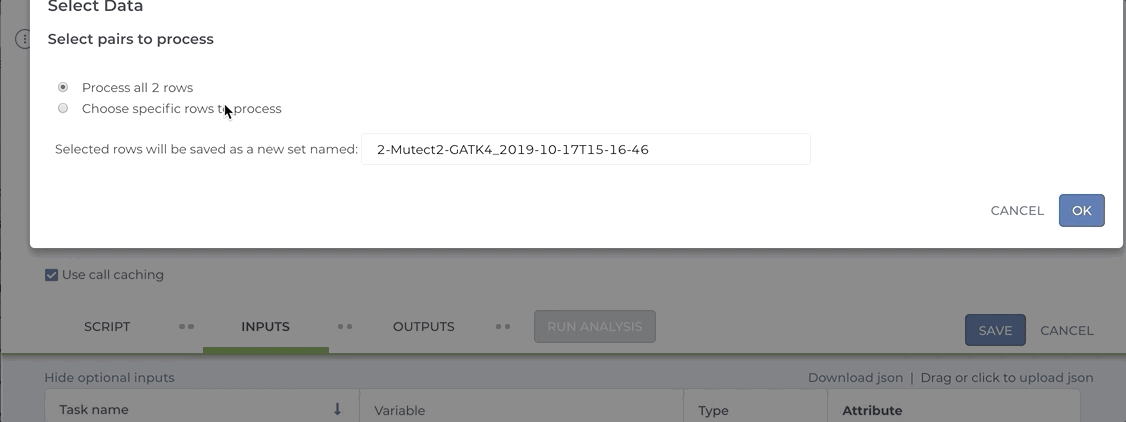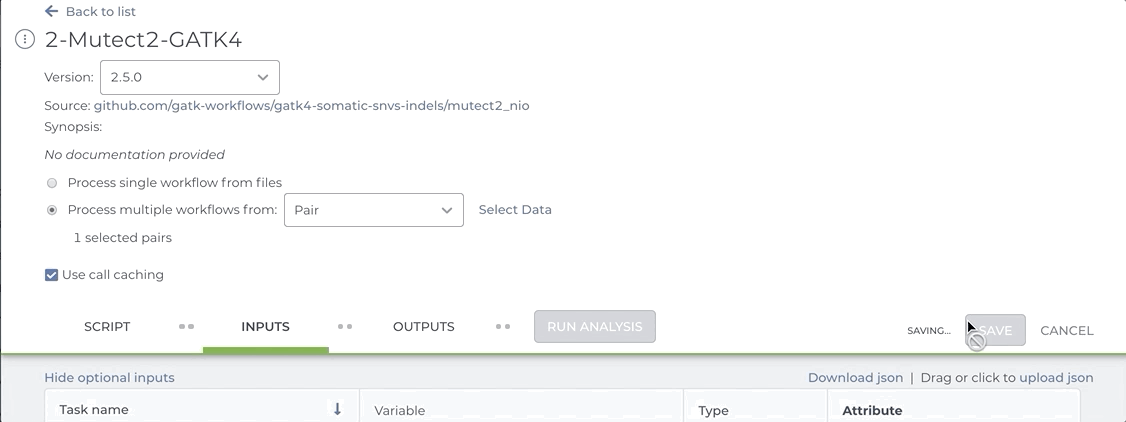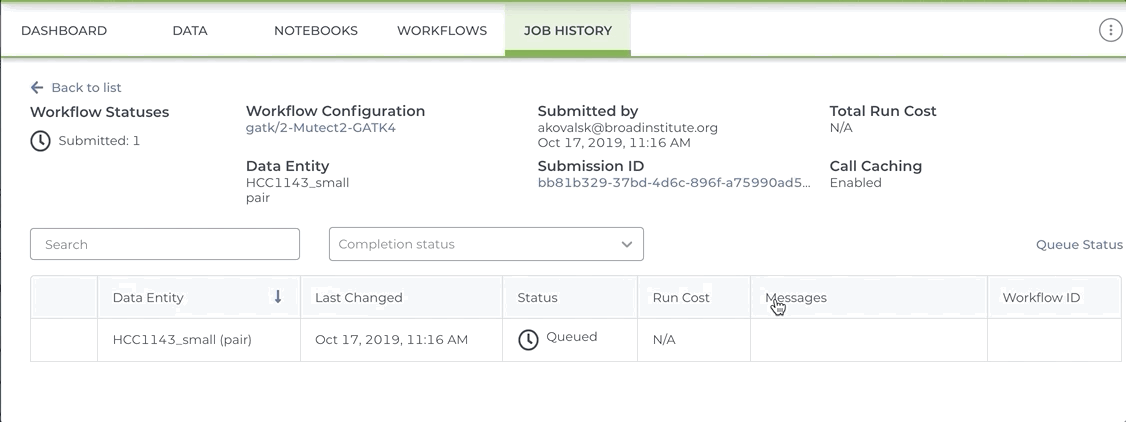
Additional resources
- For a detailed explanation of how to configure your workflow inputs for different entity and table types in the Terra interface, see Configuring workflow inputs - sets and pairs tables.
- To learn more about scripting in WDL, you can read our WDL user guide or check out the OpenWDL community - formed to steward the WDL language specification and advocate its adoption.
- Learn how to use a template to add data in a table to your workspace in this article.
- Learn how to modify, delete, and create tables in this article.
- For hands-on practice using and creating data tables try the Data QuickStart workspace.
- To learn to use data tables for input to a workflow see Workflow setup part 2: Inputs and outputs.